ARJ 2017, 1(1), P.P. 23-41;
archives/2017/sabah/
Review Article
Non-Steroidal Anti-Inflammatory Drugs Review
Sabah Jawad Salih, Ph. D. Pharm. Chem; NCDCR, Baghdad, Iraq.*
Introduction
The nonsteroidal anti-inflammatory drugs (NSAIDs) are among the most often prescribed drugs in the world. This heterogeneous class of drugs includes aspirin and several other selective or non-selective cyclooxygenase (COX) inhibitors. The non-selective NSAIDs are the oldest ones and are called traditional or conventional NSAIDs. The selective NSAIDs are called COX-2 inhibitors1.
The two main adverse drug reactions associated with NSAIDs relate to gastrointestinal effects and renal effects of the agents. These effects are dose-dependent, and in many cases severe enough to pose the risk of ulcer perforation, upper gastrointestinal bleeding, and death, limiting the use of NSAID therapy2.
NSAIDs are usually indicated for the treatment of acute or chronic conditions where pain and inflammation are present. Research continues into their potential for prevention of colorectal cancer, and cardiovascular disease. NSAIDs are generally indicated for the symptomatic relief of rheumatoid arthritis, osteoarthritis, inflammatory arthropathies, acute gout, dysmenorrhoea (menstrual pain), metastatic bone pain, headache and migraine, postoperative pain, mild-to-moderate pain due to inflammation and tissue injury, Pyrexia (fever) and Ileus3.
NSAIDs inhibit both the cyclooxygenase-1 (COX-1) and cyclooxygenase-2 (COX-2) isoenzymes. COX catalyzes the formation of prostaglandins and thromboxane from arachidonic acid (AA)4.
There is no clear-cut division between biological function of COX-1 and COX-2. Experimental evidence indicates that a full inflammation response is likely sustained by prostanoids generated by both enzymes. In this sense, drugs inhibiting both enzymes are theoretically more effective in inflammatory disease treatment. Moreover, COX-2 selective inhibitors may theoretically lead to problem in thrombosis, salt and water balance and healing. With all these aspects considered, developing of new drugs that preferentially inhibit COX-2 with moderate selectivity may be more promising5.
Mode of action of non steroidal anti-inflammatory drugs:
In 1971, Vane discovers that NSAIDs could inhibit prostaglandin synthesis and proposed that this mechanism was the basis for their pharmacological action6
Prostaglandins (PGs) are family of chemical messenger (Figure 1) which is involving in local signal within tissue; its types are known as:
PGE2: they regulate much physiological function in gut as mucosal protection, GI secretion and motility. Also responsible for triggering the hypothalamus to increase body temperature during inflammation7:
PGF2: cause myometrial contraction in humane, so use for induction of labor8:
PGI2: it is a vital vasodilator and platelet aggregation inhibitor, and the major inflammatory mediator particular in rheumatoid arthritis9:
The rate limiting step in the prostaglandins biosynthesis is the conversion of arachidonic acid to prostaglandin H2 (PGH2), in a reaction catalyzed by cyclooxygenase (COX) enzyme10.
NSAIDs inhibit prostaglandins synthesis by various effect on cyclooxygenase (COX) enzyme including non selective inhibition of COX isoform e.g. by aspirin11,or selective inhibition e.g. by etoricoxib12.
NSAIDs also are known to reduce production of super oxide radicals, induce apoptosis, inhibit the expression of adhesion molecules, decrease nitric oxide synthesis, decrease pro-inflammatory cytokines (e.g. TNF-a, IL-1), modify lymphocyte activity, and alter cellular membrane functions. However, there are different opinions as to whether these actions might contribute to the anti-inflammatory activity of NSAIDs at concentrations attained during treatment13.
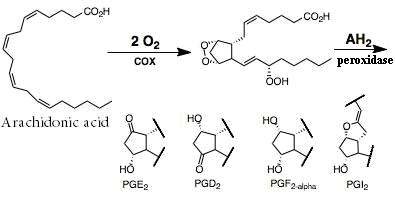
Figure 1: PGs synthesis.
Cyclooxygenases (COXs):
Cyclooxygenase is a rate–limiting enzyme for PG production14. There are at least two isoform of COX namely COX-1 and COX-2 have been identified15. which considered as a significant pharmacological target due to their various pathophysiological effects16.
Despite the structure identity of these two enzymes, and catalyze the same biochemical reaction but they are clearly different in term of amino acid sequence, tissue distribution, and physiological function, the active center of COX-2 is characterized by large pocket which can accommodate molecules with bulkier side chain than COX-117.As shown in Figure 2.
A- Cyclooxygenase-1: is a "housekeeping" enzyme expressed constitutively in many tissues, and PGs produced by COX-1 mediate vital functions such as cytoprotection of gastric mucosa18 through stimulation the synthesis and secretion of mucus and bicarbonate, and increase mucosal blood flow, in addition to its role in regulation of renal blood flow19. Also the presence of COX-1 in platelet lead to thromboxane A2 (TXA2) production that causes platelet aggregation, so it is useful in prevention of inappropriate bleeding20:
B- Cyclooxygenase-2: is an inducible enzyme, rapidly expressed in several cell types in response to growth factors, cytokines, and pro inflammatory molecules and has emerged as the isoform primarily responsible for prostaniod production in acute and chronic inflammatory conditions21. During inflammatory pain condition like rheumatoid arthritis, osteoarthritis there is significant elevation in COX-2 level in periphery and central nervous system, and this is a good scientific evidence for management of inflammation and pain with NSAIDs22. In addition to induction expression of COX-2 at sites of inflammation, several investigation have been reported constitutively expression of this isoform in several tissue and organs such as brain, kidney, and pancreas23:
C- Cyclooxygenase-3: is a novel COX splice variant, it is completely different in amino acid sequence than COX-1 and COX-2 and without COX activity, but it is involved in prostaglandins –mediated pain and inflammation24. COX-3 is important in explanation of the antipyretic and analgesic effect of paracetamol22.
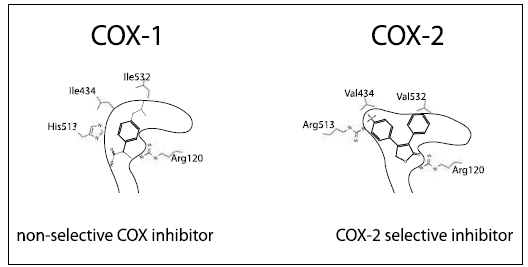
Figure 2: Difference between COX-1 and COX-2 is in size of active center25.
Therapeutic Actions of NSAIDs:
NSAIDs have three major pharmacological desirable actions. All of which result mainly from the inhibition of COX-2 in inflammatory cells and the resultant decrease in prostanoid synthesis; they are:
An anti-inflammatory action: the decrease in vasodilator PGs (PGE2, prostacyclin) means less vasodilation and, indirectly, less edema13.
An antipyretic action: NSAIDs reduce the body temperature in febrile states. The fact that selective COX-2 inhibitors are effective antipyretic agents indicates that the COX-2 predominantly involved in thermoregulation13.
An analgesic action: by decreasing PGE2 synthesis, NSAIDs repress the sensation of pain of low to moderate intensity arising from integumental structures rather than that arising from the viscera26.
Further benefits from NSAIDs are being explored, including the prevention of Alzheimer's dementia and colorectal carcinoma27. Recent studies suggest that PGs obtained via the COX-2 pathway may play a vital role in the maintenance of tumor viability, growth and metastasis28. Therefore, epidemiological evidence suggests that COX-2 inhibitors may have important therapeutic relevance in the prevention of some cancers29.
Numerous studies have demonstrated the overexpression of cyclooxygenase-2 (COX-2) in solid malignancies. Epidemiological, clinical, and preclinical investigations also provide compelling evidence that COX-2 inhibitors could act as chemopreventive agents. The anti-cancer effects of COX-2 inhibitors are based on the assumption that prostaglandins generated by COX-2 promote tumor growth in an autocrine and/or paracrine manner30.
Theoretically, COX-2 inhibitors exhibit all the anti-cancer or cancer preventive activity by blocking COX-2, thereby decrease the concentration of prostaglandins inside the tumor. However, these small molecules may also target other growth pathway, which may lead to cell growth inhibition, apoptosis or necrosis31.
Many COX-2 inhibitors can suppress the growth of non-COX-2 expressing tumor cells, while supplementation with exogenous prostaglandin cannot rescue the cells from growth inhibition caused by COX-2 inhibitors. Therefore, it is speculated that COX-2- independent effects may contribute to or even be fully responsible for the anti-cancer properties of some COX-2 inhibitors. Furthermore, the relative potency of COX-2 inhibitors to inhibit COX-2 enzyme does not match their potency to inhibit cancer cell growth32.
In addition to the lack of correlation between COX-2 inhibition and anti-cancer activities, the required concentrations of these COX-2 inhibitors to inhibit tumor cell growth significantly exceed the concentrations required to inhibit COX-2. This phenomenon suggests that the COX-2 inhibitors mainly target other pathways, which need much higher concentration forCOX-2 inhibitors to block33.
The strongest evidence for a COX-independent mechanism is that some non-COX-2 inhibitory derivatives of certain COX-2 inhibitors still exhibit significant anti-cancer activity34.
COX-1: COX-2 Selectivity:
Selectivity is expressed as a ratio of the IC50, of a particular NSAID against each respective isoenzyme. Most assay results are reported as the IC50 of thromboxane B2 (TXB2) for COX-1 inhibition and IC50 of PGE2 for COX-2 inhibition. The IC50 is the amount of drug necessary to inhibit COX-1 or COX-2 activity by 50% and is determined using various above assays. The higher the IC50, the more drug necessary to inhibit the particular enzyme. Therefore, a COX-1: COX-2 ratio greater than 1 would indicate more drug is necessary to inhibit COX-1 than COX-2 and that drug would selectively inhibit COX-2 and spare COX-133.
Considerations Regarding the Pharmacology of COX-Inhibitors:
Several important considerations should not be overlooked in the discussion of the pharmacology of COX-inhibitors:
- The relationship between the relative inhibition of COX-1 and COX-2 and alteration of PG-mediated biological functions is not linear35.
- As pharmacological targets, the dose effect thresholds of efficacy and safety for COX-1 and COX-2 inhibition are probably indefinable. Even if it was possible to accurately predict the relative selectivity of COX inhibition in vivo, it is still not known to what extent, and for how long, COX-1 can be inhibited without an increased risk of GI toxicity. Conversely, the degree of COX-2 inhibition needed to produce anti-inflammatory responses in vivo also is unknown36.
- There are currently insufficient data to accurately correlate biochemical and pharmacological measures of COX selectivity with clinical efficacy and safety37.
Structural Properties of COX-1 and COX-2 for Substrate and Inhibitor Binding:
The amino acid sequence of both enzymes is closely related and the structures are very similar. COX-1 and COX-2 enzymes are homodimers; each monomer consists of three sites, an epidermal growth factor-like domain, a membrane-binding domain and a catalytic domain that contains both the COX and peroxidase active sites38.
Both COX-1 and COX-2 are associated with the membrane and each consists of a long channel with a bend at the end, the channel being wider in COX-2 as shown in figure (2)39.
These two enzymes share sixty-percent homology in amino acid sequence. However, the conformation for the substrate-binding site and catalytic regions are slightly different. Eight amino acid residues play an important role for the substrate and inhibitor binding in the COX-channel [the amino acid numbering refers to the COX-1 and COX-2 enzyme coding (for the residues in COX-2 one has to subtract 14 to reach the homologous amino acid residue in COX-1)]38.
Active site (Catalytic Center):
The amino acid tyrosin 385 (Tyr385) in COX-1 (Tyr371 in COX-2) is located at the top of the channel and represent together with the heme group the catalytic center39.
Acylation Site:
In the structure of COX-1, aspirin acetylates serine 530 (Ser530) (Ser516 in COX-2) preventing the binding of arachidonic acid to active site of the enzyme. X-ray analysis has elucidated an additional function of Ser530. This polar amino acid is involved in the binding of inhibitors with a benzyol group, such as indomethacin, or with an anilino NH, such as diclofenac and meclofenamate40.
Side Pocket and Extra Space in COX-2:
The crucial difference between the two COX enzymes is at position 523: here COX-1 has a bulky isolucine amino acid while COX-2 has a valine residue (Val509) a smaller molecule that leaves a gap which gives access to a "side-pocket". Additionally, the amino acid exchange of histidine 513 (His513) in COX-1 for Argenine 499 (Arg499) in COX-2 allows a hydrogen bonding with the sulfon part of COX-2 inhibitors40.
Also the amino acid phenylalanine 503 (Phe503) in COX-1 is replaced by leucine 489 (Leu489) in COX-2. This smaller residue in COX-2 allows the first shell amino acid Leu384 to create an "extra space" at the top of the binding site in COX-2, thus allowing larger inhibitors to bind38.
Ionic Binding:
Arachidonic acid (substrate) and acidic COX-inhibitors are bind via their carboxylate anion to the guanidinium cation of Arg120 (Arg106 in COX-2). This has been shown by X-ray analysis for COX binding for arachidonic acid flubiprofen and indomethacin39,40.
H-bonding Dynamics:
Arg120 and His513 in COX-1 (Arg106 and Arg499 in COX-2) are involved together with Tyr355 and glutamic acid 524 (Tyr341 andGlu510 in COX-2) in a hydrogen bonding networks. This has been postulated from X-ray analysis41.
The two H-bonding networks are proposed to be responsible for allosteric activation of the COX enzyme (Figure 3), and this is the best structural explanation for the time-dependency of COX-2 inhibition and of the loss of COX-1 activity due to the need of allosteric enzyme activation of COX-2 selective NSAIDs42.

Figure 3: Different ligands bind either the allosteric or the catalytic subunit. Allosteric subunit binds a non-substrate, activating FA (e.g., palmitic acid). The allosteric subunit with bound fatty acid activates the catalytic subunit by decreasing the Km for AA43.
Chemical Categories of Traditional NSAIDs:
The NSAIDs are a group of chemically dissimilar agents that differ in their antipyretic, analgesic and anti-inflammatory activities27. These drugs are belonging to the following various chemical categories:
- Salicylic Acid Derivatives: Aspirin.
- N-Arylanthranilic Acid Derivatives (Fenamates): Mefenamic acid.
- Enolic Acids (Oxicams): Piroxicam, Tenoxicam
- 4. Heteroaryl Acetic Acid and Aryl Acetic Acid Derivatives: Indomethacin, Sulindac, Benzene acetic acid, Diclofenac, Tolmetin sodium and Naproxen (Figure 4) which has a longer half-life than most of the other structurally and functionally similar agents making twice-daily administration feasible. It approximately twenty times more potent than aspirin as cyclooxygenase inhibitor13.
Figure 4: Naproxen
COX-2 Selective Inhibitors:
COX-2 selective inhibitors or coxibs were developed to inhibit prostacyclin synthesis by the COX-2 isoenzyme induced at the site of inflammation without affecting the action of the constitutively active COX-1 isoenzyme found in the gastrointestinal tract, kidneys and platelets44.
The International Consensus Meeting on the Mode of Action of COX-2 Inhibitors (ICMMAC) provided a definition of COX-2 specificity as: these compounds are a new class of drugs that specifically inhibit the enzyme COX-2 while having no effect on COX-1 across the completely therapeutic dose range45.
The main question regarding selective COX-2 inhibitors, as to whether are really better tolerated, or only have (new) side effects different from those of standard NSAIDs, will be answered by epidemiological data in the future. Despite this unclear situation, medicinal chemists have done their job well. Many hundareds of COX-2 inhibitors have been described46.
The structural difference between COX-1 and COX-2 has allowed the development of COX-2 selective agents that differ from most of the traditional NSAIDs, which inhibit both COX-1 and COX-227. However, preferential COX-2 inhibitors were typically developed before the existence of COX-2 was known. These compounds represent examples where potent anti-inflammatory activity was seen in standard inflammatory models with less ulcerogenic effects than standard NSAIDs most notably meloxicam47.
Common Pharmacophore for Selective COX-2 Inhibitors:
There have been remarkable efforts concerning the identification of selective COX-2 inhibitors with an attractive pharmacological profile. Five structural classes can be identified with additional class bearing little or no resemblance to one another in their molecular structure. Most of these compounds showed competitive time-dependent inactivation of COX-2 but no time dependency in context with COX-1. These inhibitors caused induced conformational changes in COX-2 enzyme by binding very tightly but non-covalently to the enzyme. This time dependent strong binding to the inducible enzyme is responsible for their observed specificity48.
Carbocycles and Heterocycles with Vicinal Aryl Moieties:
The greatest research activities in the field of COX-2 inhibitors have been made in the synthesis and pharmacological testing of this class of compounds. This structural class is characterized by a central carbocyclic or heterocyclic ring that bears two phenyl substituents attached at the vicinal positions. A wide variety of heterocycles can serve as a template for COX-2 selective inhibitors. In accordance with current data one aromatic ring must be substituted with methylsulfonyl or sulfonamide substitute in the para-position for COX-2 selectivity. Decreased COX-2 specificity but improved oral bioavailability is observed by replacing the methylsulfone with the sulfonamide moiety. By far most of these compounds bear a halogen atom such as fluoro or chloro at the second phenyl ring49.
The pyrazole derivative, celecoxib (Figure 5) has been launched for the treatment of rheumatoid arthritis. It is as effective as other NSAIDs in rheumatoid arthritis and osteoarthritis, and in trials it has caused fewer endoscopic ulcers than most other NSAIDs37.
Figure 5: celecoxib
The furanone derivative rofecoxib (Figure 6a) is significantly more selective as COX-2 inhibitor than celecoxib with the ratio of 267:30 respectively. This high selectivity of rofecoxib is expected to disrupt the balance between antithrombotic prostacyclin and prothrombotic thromboxane and it’s the basis for the adverse cardiovascular events. In a study, it was shown that the incorporation of a para-N-acetyl sulfonamido substitute on the phenyl ring of the rofecoxib regio isomer (Figure 1-6b) provided a highly potent and selective COX-2 inhibitor that has the potential to acetylate the COX-2 isozyme50.
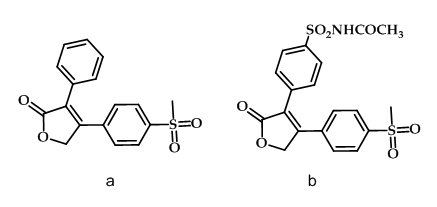
Figure 6: Rofecoxib & its derivative
Valdecoxib (Figure 7a), a diaryl-substituted isoxazole, is as effective as non-selective NSAIDs for treatment of rheumatoid arthritis with no effect on platelet aggregation or bleeding time50, therefore caused an increased number of adverse cardiovascular events when used for pain management in coronary artery bypass surgery51. N-acylation of Valdecoxib served the dual role of acylation agent and prodrug as illustrated in paracoxib (Figure 7b) which was found to be selective COX-2 inhibitor51.
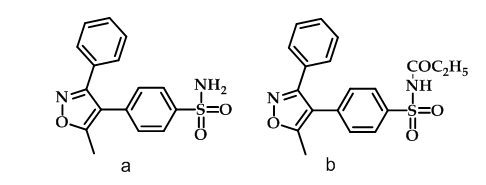
Figure 7: Valdecoxib & paracoxib
The substitution pattern on the heterocyclic ring is also important for the efficacy as demonstrated in the series of bromo-substituted thiophene derivatives as selective COX-2 inhibitors with the 5-bromothiophene derivative (Figure 8) with the code name (Dup-697) being the most potent compound in acute and chronic anti-inflammatory in vivo models with high selectivity52.
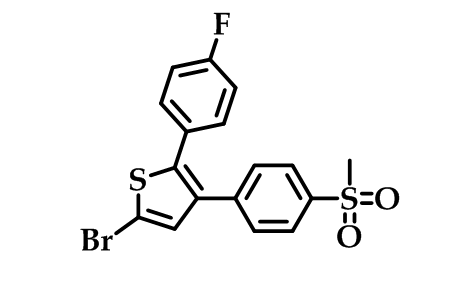
Figure 8: Dup-697
Diaryl- or Aryl/Heteroaryl Ether and Thioether Derivatives:
Nimesulide (Figure 9a) is a well known analgesic, and flosulide (Figure 9b) is an older member of this class. Both were found to be selective COX-2 inhibitors. By replacing the oxygen atom of flosulide with a sulfur atom (Figure 9c) increased anti-inflammatory potency and better gastrointestinal safety was observed. Interestingly, the in vitro activities of flosulide and the sulfur analogue against human COX-1 and COX-2 were identical but the sulfur compound was shown to possess greater oral bioavailability. An electron-withdrawing substituent at the aromatic ring seems to be essential for physiological activity with an optimum for the cyano and the acetyl group53.
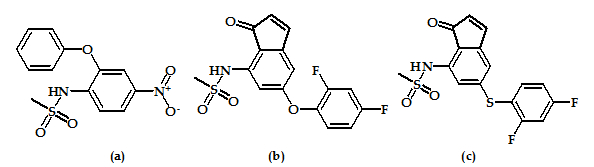
Figure 9: Nimesulide, flosulide & flosulide derivative.
Cis-Stilbene Derivatives:
The invention encompasses the novel stilbene analogous compounds of the formula shown in (Figure 10) were useful in the treatment of COX-2 mediated disease.
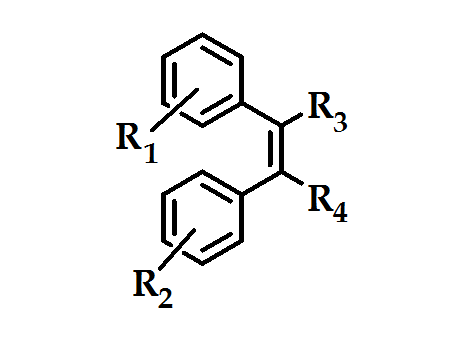
Figure 10: stilbene analogous compounds
The methylsulfone moieties R1 in combination with a halogen atom R2 were advantageous for COX-2 selectivity, with much greater structural variety of the substituents R3 and R4. These compounds were produgs, by virtue of their in vivo conversion to compounds with high inhibitory activity against COX-2 and/or specificity for COX-2 over COX-1 (Figure 11). These agents will prove to be useful as an alternative to conventional NSAIDs, particularly where such NSAIDs may be contraindicated. However, according to patent applications, all derivatives are only in an early stage of development (biological testing)54.
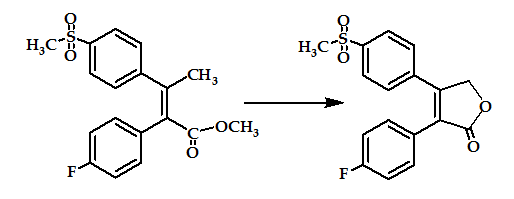
Figure 11: stilbene analogous prodrug.
Diaryl and Aryl/Heteroaryl Ketones:
The ketone function, as a link between two aryl or an aryl ring and a heterocycle, is long known in the class of anti-inflammatory drugs, i.e. as nonspecific inhibitors of cyclooxygenases, such as tolmetin (Figure 12a) and zomepirac (Figure 12b). The ratio of selectivity in favor of COX-2 was achieved by substitution of the acetic acid group of zomepirac by an oxopyridazinyl moiety at the pyrrole ring as in compound (Figure 12c) which is highly selective COX-2 inhibitor.
Other selective COX-2 inhibitors were obtained by different structural modifications of the substituents at both rings. However, all derivatives are only in an early stage of development (biological testing) or pre-clinical study55.
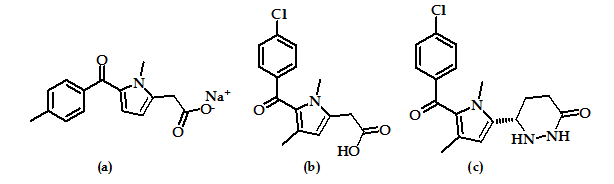
Figure 12: Tolmetin, Zomepirac and Zomepirac derivative.
Benzylidene-Heterocycles Derivatives:
A variety of benzylidene oxazoles-thiazoles, and imidazoles with the formula shown in (Figure 13), has been prepared and evaluated as COX-2 inhibitors.
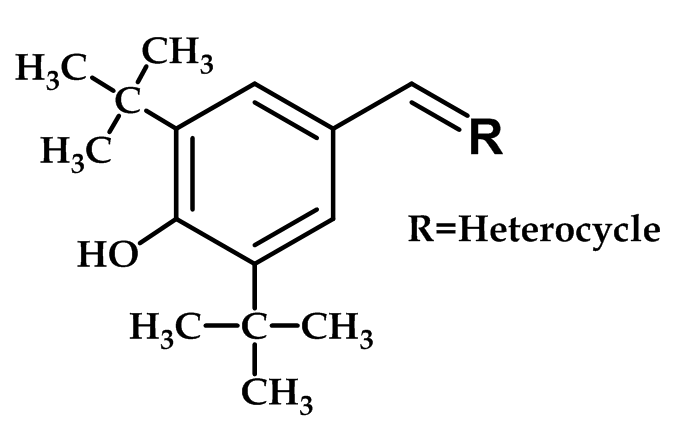
Figure 13: Benzylidene-Heterocycles Derivatives
Thiozole and oxazole derivatives with the 2-hydroxy, 2-mercapto or 2-imino substituent linked to the di-tert-butylphenol derivative were found to be potent and selective COX-2 inhibitors. The potency and selectivity are extremely sensitive to minor changes in chemical structure within this chemical series; with phenolic OH is essential for potency. However, all derivatives are only in an early stage of development (biological testing) or pre-clinical study56.
Structural Variations of Known NSAIDs and Compounds without Structural Features:
An increasing number of research indicated that structural modifications of commercially available nonselective NSAIDs improved the specificity for COX-2. Aspirin is the only known NSAID that covalently modifies both COX-1 and COX-2, but it is a 10- to 100-fold more potent inhibitor of COX-1 relative to COX-257. In accordance with the known ionic interaction between the carboxylate of aspirin and the argenine residue adjacent to Ser530, the carboxylate moiety in aspirin was replaced with the methyl sulfide and this derivative was identified as a lead compound that demonstrated moderate inhibitory potency and selectivity for COX-258.
Systematic structural modifications led to the development of compound [O-(acetoxyphenyl)-hept-2-ynyl sulfide] (Figure 14) which was the most potent and selective inhibitor in the series. This derivative was found to be 60 times more active against COX-2 than aspirin and its selective inhibition toward COX-2 was resulted from the acetylation of the same serine residue that aspirin acetylates. This compound also is claimed to be superior to celecoxib in inhibiting cell growth of colorectal carcinoma cells59.
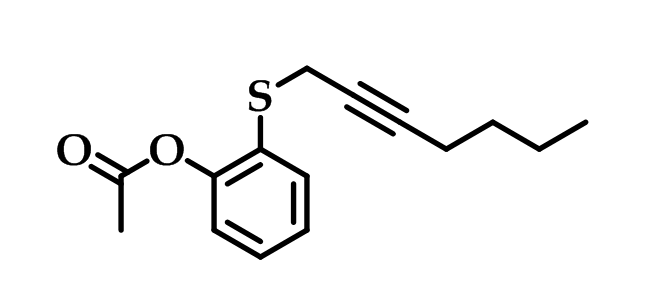
Figure 14: O-(acetoxyphenyl)-hept-2-ynyl sulfide
Replacement of the N-p-chlorobenzoyl group of indomethacin (Figure 15a) by a 2,4,6-trichlorobenzoyl unit removed the COX-1 inhibitory activity and shifted it to COX-2 selectivity as in compound (Figure 15b)60. The highest COX-2 selectivity was found when the chlorobenzoyl moiety is replaced with 4-bromobenzyl group as in compound (Figure 15c)61, in addition to change in the acidic side-chain at 3-poistion as in compounds (Figure 15d) and (Figure 15e)62.
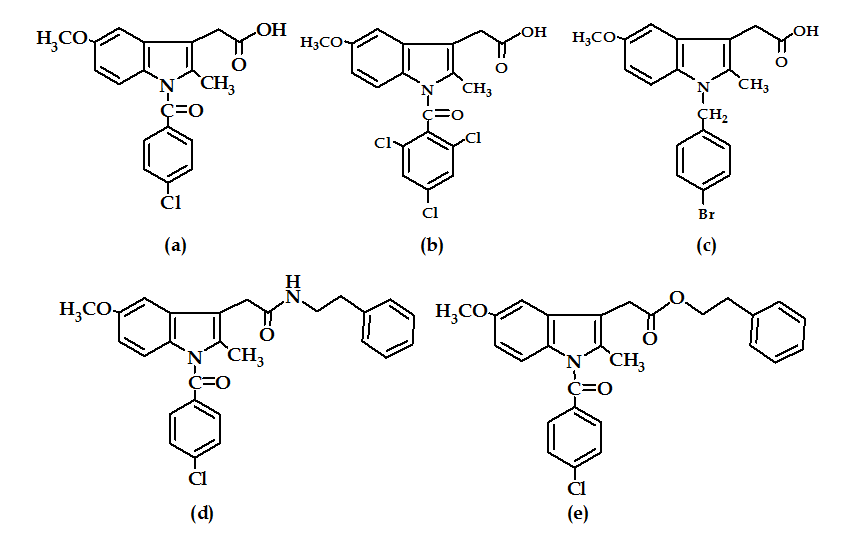
Figure 15: Indomethacin and its derivatives
It has been found that the IC50 of COX-1: COX-2 of compound (Figure 15c) was about 26; while that for compounds (Figure 15d and e) were about 1100 and 1320 respectively. Therefore, the effect of the nature of the side chain at 3-position is more significant on the selectivity than the nature of halogen atom63.
Since the discovery, enol-craboxamide bears some selective inhibitory activity such as meloxicam (Figure 16a), this class of compounds attached more attention. SAR of enol-carboxamide type NSAIDs indicated that the N-methyl was essential for activity in meloxicam-like derivatives, but a benzyl substituent was tolerated also. Modification of these compounds gave rise to compound (Figure 16b) which is a COX-2 selective inhibitor with anti-inflammatory activity in vivo63.
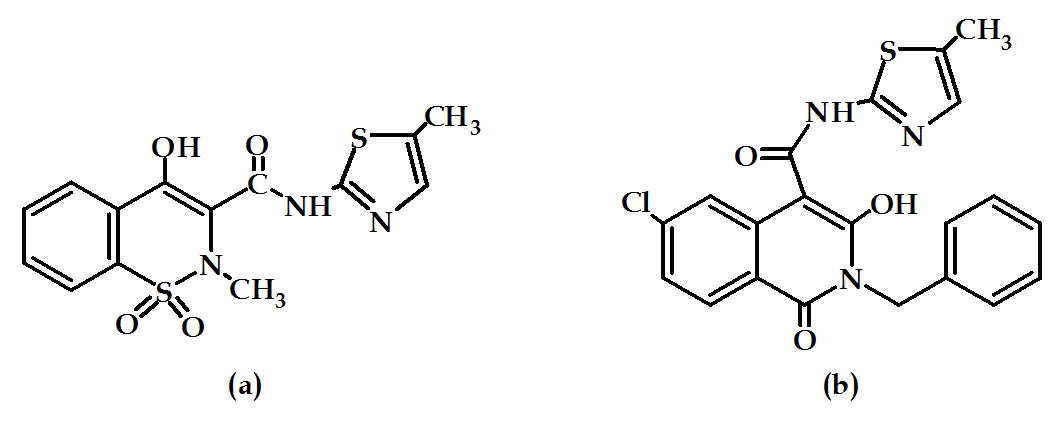
Figure 16: Meloxicam and its derivative.
An example of compounds with high COX-2 selectivity but without common structural features is compound (Figure 17) which may be interpreted as a special case of chemical structures belong to heterocycles with vicinal aryl moieties in which the fluorine atom and the sulfonamide moiety are attached at the same benzene ring. In addition, one phenyl ring is replaced by a cyclohexyl group64.
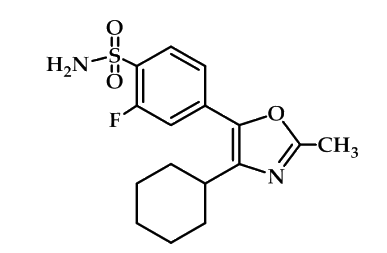
Figure 17: heterocycles with vicinal aryl moieties
The aryl-sulfonamide derivatives such as compound (Figure 18) were found to be more active than indomethacin and nimesulide at the same molar concentration as anti-inflammatory agents with high COX-2 selectivity65.

Figure 18: Aryl-sulfonamide derivatives
Finally, the 2, 3-dihydro-3, 3-dimethylbenzofuran derivatives like compound shown in Figure 19, are inhibitors of cyclooxygenase with selectivity for the COX-2 isoform. Changes in the degree of substitution, ring size, and heteroatom identity are all tolerated to varying degrees. These compounds are potent inhibitors of COX with up to 33-fold selectivity for COX-266.
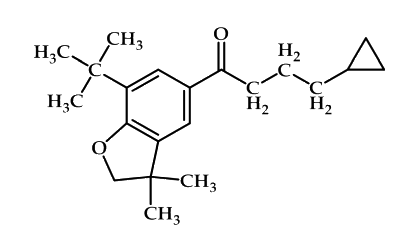
Figure 19: 2, 3-dihydro-3, 3-dimethylbenzofuran derivatives
References
1 Howard, Patricia A: Nonsteroidal anti-inflamatory drugs and cardiovascular Risk. J. Am. Coll. Cardiol, 2004, 43: pp.519-25.
2 Green, Ga: Understanding NSAIDs: from aspirin to COX-2, Clinical cornerstone, 2001, 3 (5): pp.50–60
3 Simone Rossi, ed; Australian medicines handbook, 2006. Adelaide: Australian Medicines Handbook Pty Ltd.
4 Chandrasekharan, Nv; Dai, H; Roos, Kl; Evanson, Nk; Tomsik, J; Elton, Ts; Simmons, Dl: COX-3, a COX-1 variant inhibited by acetaminophen and other analgesic/antipyretic drugs: cloning, structure, and expression, Proceedings of the National Academy of Sciences of the United States of America, 2002, 99 (21): pp.13926–31.
5 Chen, X.H.; et al: Imrecoxib : a novel and selective COX-2 inhibitor with anti-inflammatory effect, Acta. Pharmacol. Sci, 2004, 25(7): pp.927-931.
6 Ahmed, S.P. and Rasheed, S.: Studies on the mode of action of non-steroidal anti-inflammatory drugs, Pakistan J. Pharm. Sci, 1989, 2(2): pp.1-11.
7 Dey,I.; Lejeune, M. and Chadee, K.: Prostaglandin E2 receptor distribution and function in the gastrointestinal tract, British J. of Pharmacology, 2006, 149: pp.611-623.
8 Dey,I.; Lejeune, M. and Chadee, K.: Prostaglandin E2 receptor distribution and function in the gastrointestinal tract, British J. of Pharmacology, 2006, 149: pp.611-623.
9 Stitham, J.; Midgett, C.; Martin, K.A, and Hwa, J.: Prostacyclin: an inflammatory paradox, Pharmacology. 2011, 2 (24): pp.1-6.
10 Kalgutkar, A.S.; Rowlinson, S.W.; Crews, B.C. and Marnett, L.J.: Amide Derivatives of Meclofenamic Acid as Selective Cyclooxygenase–2 Inhibitors, Bioorganic & Medicinal Chemistry Letters, 2002, 12: pp.521 –524.
11 Suneela, S.D.; Mini, K.; Badal, R. and Kadam, S.S.: Synthesis, kinetic studies and pharmacological evaluation of mutual azo prodrug of 5- amino salicylic acid for colon - specific drug delivery in inflammatory bowel disease, Eur. J. of Med. Chem, 2007, 42: pp.885-890.
12 Afshin, Z.; Leila, N.; Bahram, D.; Orkideh, G.D. and Mehdi, H.: Synthesis of 2, 3-diaryl-1, 3-thiazolidine-4-one derivatives as selective cyclooxygenase (COX-2) inhibitors, Bioorganic & Medicinal Chemistry Letters, 2007, 17: pp.5634–5637.
13 Laurence, L.B.; John, S.L. and Keith, L.P.: Goodman and Gilman's, The Pharmacological Basic of Therapeutics, 11th ed, 2008, The McGraw-Hill, Inc U.S.A, p. 429.
14 Hayashi, S.; Ueno, N.; Murase, A.; Nakagawa, Y. and Takada, J.: Novel acid- type cyclooxygenase -2 inhibitors: Design, Synthesis, and Structure-activity relationship for anti-inflammatory drug, Eur. J. of Med. Chem, 2012, 50: pp.179-195.
15 Guo, J.S.; Cho, C.H.; Wang, J.Y. and Koo, M.W.L.: Differential effects of selective and non – selective inhibition of nitric oxide synthase on the expression and activity of cyclooxygenase – 2 during gastric ulcer healing, Eur. J. of Pharm, 2006; 536: pp.301–308.
16 Hayashi, S.; Sumi, Y.; Ueno, N.; Murase, A; Takada, J.: Discovery of a novel COX-2 inhibitor as an orally potent anti-pyretic and anti- inflammatory drug: Design, synthesis, and structure–activity relationship, Biochemical Pharmacology, 2011, 82: pp.755–768.
17 Lee, B.; Kwak, J.H.; Huang , S.W.; Jang, J.Y.; Lim, S.; Kwak, Y.S.; Lee, K.; Kim, H. S.; Han, S. B.; Hong, J. T.; Lee, H.; Song, S.; Seo, S. Y.; Jung, J. K.: Design and synthesis of 4-O-methylhonokiol analogs as inhibitors of cyclooxygenase -2 (COX-2) and PGF production, Bioorganic and Medicinal Chemistry, 2012, 1;20(9): pp.2860-2868.
18 Arfaie, S; Zarghi, A: Design, synthesis and biological evaluation of new (E) – and (Z)-1, 2, 3-triaryl-2-propen -1- ones as selective COX -2 inhibitors, Eur. J. of Med. Chem, 2010, 45: pp.4013- 4017.
19 Parente, L; Perretti, M: Advance in pathophysioloyg of constitutive and inducible cyclooxygenases: two enzyme in the spotlight, Biochemical pharmacology, 2003, 65: pp.153-159.
20 Botting, R.M.: Inhibitors of cyclooxygenases: mechanisms, selectivity and uses, J. of physiology and pharmacology, 2006, 57(5): 113-124.
21 Minghetti, L.: Cyclooxygenase -2 (COX-2) in Inflammatory and Degenerative Brain Diseases, J. of Neuropathology and Experimental Neurology, 2004, 63 (9): pp.901- 910.
22 Baliki, M.; Katz, J.; Chialvo, D.R.; Apkarian, A.V.: Single subject pharmacological -MRI (ph MRI) study: Modulation of brain activity of psoriatic arthritis pain by cyclooxygenase-2 inhibitor, Molecular Pain, 2005, 1(3): pp.1-8.
23 Porcher, C.; Horowitz, B.; Bayguinov, O.; Ward, S.M.; Sanders, K.M.: Constitutive Expression and Function of Cyclooxygenase -2 in Murine Gastric Muscles, Gastroenterology, 2002, 122: pp.1442-1454.
24 Kis, B.; James, A. Snipes, and Busija, D.W.: Acetaminophen and the Cyclooxygenase -3 Puzzle: Sorting out Facts, Fictions, and Uncertainties, J. of pharmacology and experimental Therapeutics, 2005, 315(1): pp.1-7.
25 Grosser T; The pharmacology of selective inhibition of COX-2, Thromb Haemost, 2006, 96: pp.393–400.
26 Harvey, R.A. and Champe, P.C. (Eds.): Lippincott’s illustrated reviews pharmacology, 5th Ed, 2012, p. 495.
27 Laurance, D.R.; Bennett, P.N. and Brown, M.J. (Eds.): Clinical pharmacology, 9th Ed, 2003, Churchill Livingstone, London, p. 280.
28 Amit, S.K; Zhao, Z.: Discovery and design of selective COX-2 inhibitors as Non-ulcerogenic, anti-inflammatory drugs with potential utility as anti-cancer agents, Curr. Drug Targets, 2001, 2, pp.79-106.
29 Mitchell, J.A; Warner, T.D.: COX-2: pharmacology, physiology, biochemistry and relevance to NSAID therapy, Br. J. Pharmacol, 1999, 128, pp.1121-1132.
30 Keller, J.J.; Giardiello, F.M.: Chemoprevention strategies using NSAIDs and COX-2 inhibitors, Cancer Biol. Ther, 2003, 2, pp.S140-S149.
31 Rao, C.V.; Reddy, B.S.: NSAIDs and chemoprevention, Curr. Cancer. Drug. Targets, 2004, 4, pp.29-42.
32 Hanif, R. ; Pittas, A.; Feng, Y.; Koutsos, M.I.; Qiao, L.; Staiano-Coico, L.; Shiff, S.I.; Rigas, B.: Effects of nonsteroidal anti-inflammatory drugs on proliferation and on induction of apoptosis in colon cancer cells by a prostaglandin- independent pathway, Biochem. Pharmacol, 1996, 52, pp.237-245.
33 Han, S.; Roman, J.: COX-2 inhibitors suppress integrin alpha5 expression in human lung carcinoma cells through activation of Erk: involvement of Sp1 and AP-1 sites, Int. J. Cancer, 2005,116, pp.536-546.
34 Zhu, J.; Huang, J.W.; Tseng, P.H.; Yang, Y.T.; Fowble, J.; Shiau, C.W.; Shaw, Y.J.; Kulp, S.K.; Chen, C.S.: From the cyclooxygenase-2 inhibitor celecoxib to a novel class of 3-phosphoinositide-dependent protein kinase-1 inhibitors, Cancer Res, 2004, 64, pp.4309-4318.
35 Patrono, C.; Patrignan, P. and Garca Rodriguez, L.A.: COX selective inhibition of prostanoid formation, J. Clin. Invest, 2001, 108: pp.7-13.
36 Lipsky, L.P.E.; Abramson, S.B.; Crofford, L: The classification of COX inhibitors, J. Rheumatol, 1998, 25: pp.2298-2303.
37 Everts, B.; Wghrborg, P. and Hender, T.: COX-2 specific inhibitors – the emergence of a new class of analgesic and anti-inflammatory drugs, Clin. Rheumatol, 2000, 19: pp.331-343.
38 Van, J.; Botting, J. (Eds.): Selective COX-2 inhibitors: Pharmacology, clinical effects and therapeutic potential, Kluwer Academic publishers, Dordrecht, 1998, pp. 19-26.
39 Rang, H.P.; Dale, M.M.; Ritter, J.M. (Eds): Pharmacology, 4th Ed, 2003, Churchill Livingstone, London, p.244.
40 Kurumbail, R.G.; Stevens, A.M.; Gierse, J.K.: Structural basis for selective inhibition of cyclooxygense-2 by anti-inflammtory agents, Nature, 1996, pp. 384, 644.
41 Luong, C.; Miller, A.; Baranett, J.; Chow, J.: Flexibility of the NSAID binding site in the structure of human cyclooxygense-2. Nat. Struct. Biol. 1996, 3: pp.927-933.
42 So, O-Y.; Scarafia, E.; Mak, A.Y.; Callan, O.H.; Swinney, D.C.: The dynamics of prostaglandin H synthase. Studies with prostaglandin H synthase-2 Y355F unmask mechanism of time dependent inhibition and allosteric activation, J. Biol. Chem, 1998, 273: p.5801.
43 Dong, L; Vecchio, AJ; Sharma, NP; Jurban, BJ; Malkowski, MG; Smith, WL: Human cyclooxygenase-2 is a sequence homodimer that functions as a conformational heterodimer, J. Biol. Chem, 2011, 286 (21): pp.19035–46.
44 Katzung, B.G. (Ed.): Basic and clinical pharmacology, 9th Ed, 2004, McGraw-Hill, New York, p. 576.
45 Brooks, P.; Evans, J.F.; Fenner, H: Interpreting the clinical significance of the differential inhibition of COX-1 and COX-2, Br. Soci. Rheumatol. 1999, 38: pp.779-788.
46 Crofford, L.J.: The role of COX-2 in health and disease. Inflammation; 4th congress, Paris, IDrugs, 1999, 2(9): pp.882-885.
47 Schassmann, A.; Peskar, B.M.; Settler, C.; Netzer, P.: Effect of inhibition of prostaglandin endoperoxide synthase-2 in chronic gastrointestinal ulcer models in rats, Br. J. Pharmacol, 1998, 123: p.795.
48 Kalgutkar, A.S.; Crews, B.C.; Marnett, L.J.: Kinetics of the interaction of nonsteroidal anti-inflammatory drugs with prostaglandin endoperoxide, Biochemistry, 1996, 35: p.9076.
49 Reitz, D.B.; Li, J.J.; Norton, M.B.: Selective cyclooxygenase inhibitors: novel 1,2-diarylcyclopentenes are potent and orally active COX-2 inhibitors, J. Med. Chem, 1994, 37: p.3878.
50 Cleland, L.G. and James, M.J.: COX-2 selectivity varies across class, Australian Health Review, Journal of Health Care Decision Makers (MJA), 2005, 182(4): pp.197-198.
51 Boyer-Joubert, C.; Lorthiois, E. and Moreau, F.: Annual reports in medicinal chemistry, Elsevier Inc, 2003, pp. 347-374.
52 Gierse, J.K.; Hauser, S.D.; Creely, D.P.: Expression and selective inhibition of the constitutive and inducible forms of human cyclo-oxygenase, Biochem. J, 1995, 305: p.479.
53 Tanaka, K.; Makino, S.; Shimotori, T.: Pharmacological studies of the new antiinflammatory agent 3-formylamino-7-methylsulfonylamino-6-phenoxy-4H-1-benzopyran-4-one, Arzneim-Forsch/Drug Res, 1992, 42: pp. 935–950.
54 Black; et al.: Diphenyl synthesis as prodrugs to COX-2 inhibitors, United State Patent, Patent number 5733909, 31/3/1998.
55 Barente, J.W.; Dunn, J.P.; Kertesz, D.J.; et al.: Combination therapy of radiation and a COX-2 inhibitor for the treatment of neoplasia, EP 1999; Patent number 714895 AI.
56 Unangst, P.C.; Connor, D.T.; Wiaczeslaw, A.; et al.: Synthesis, structure-activity relationships, and in vivo evaluations of substituted di-tert-butylphenols as a novel class of potent, selective, and orally active COX-2 inhibitors, J Med Chem, 1999, 8, 42(7): pp.1161-1169.
57 Meade,E.A.;Smith,W.L.;De Witt,D.L.;et al.: Differential inhibition of prostaglandin endoperoxide synthase (cyclooxygenase) isoenzymes by aspirin and other NSAIDs, J Biol.Chem, 1993, 268, p.6610.
58 Kalgutkar,A.S.; Kozak,K.R.; Crews,B.C.;et al: Covalent modification of COX-2 by 2-acetoxyphenylenyl alkyl sulfides, a new class of selective COX-2 inactivators, J.Med.Chem, 1998, 41, p.4800.
59 Supuran, C. T: 2-(Acetoxyphenyl)-(Z)-styryl sulfides as cyclooxygenase inhibitors, Expert Opinion on Therapeutic Patents, 2001, 11(7), pp.1223-1227.
60 Black, W.C.; Bayly, C.; Belley, M.; et al: From indomethacin to a selective COX-2 inhibitor: development of indolalkanoic acids as potent and selective COX-2 inhibitors, Bioorg.Med.Chem.Lett, 1996, 6, p.725.
61 Leblanc, Y.; Black, W.C.; Chan, C.C.; et al.: Synthesis and biological evaluation of both enantiomers of L-761,000 as inhibitors of COX-1 and 2, Bioorg. Med. Chem. Lett, 1996, 6, p.731.
62 Kalgutkar AS; Crews BC; Rowlinson SW; Marnett AB; Kozak KR; Remmel RP; Marnett LJ.: Biochemically based design of cyclooxygenase-2 (COX-2) inhibitors: facile conversion of nonsteroidal antiinflammatory drugs to potent and highly selective COX-2 inhibitors, Proc Natl Acad Sci U S A, 2000, 18; 97(2), pp.925-930.
63 Lazer, E.S.; Sorcek, R.; Cywin, C.L.; et al.: Anti-inflammatory 2-benzyl-4-sulfonyl-4-H-isoquinoline-1,3-diones: novel inhibitors of COX-2. Bioorg. Chem. Lett, 1998, 8, p.1181.
64 Matsushita, M.; Masaki, M.; Yagi, Y.; et al.: Pharmacological profile of JTE-522, a novel prostaglandin H synthase-2 inhibitors, in rats, Inflamm. Res, 1997, 46, pp.461-466.
65 Lages, A.S.; Silva, K.C.M.; Miranda, A.L.P.; et al.: Synthesis and pharmacological evaluation of new flosulide analogues, synthesized from natural safrole, Bioorg. Chem. Lett, 1998, 8, p.183.
66 Fernández-Forner, D.; Guardiola M.: Drug data report: Anti-inflammatory drugs: 2,3-dihydro-3,3-dimethylbenzofuran derivatives, 1998, 20(3), pp.197-280.
© 2017 by the authors; licensee ARJ